


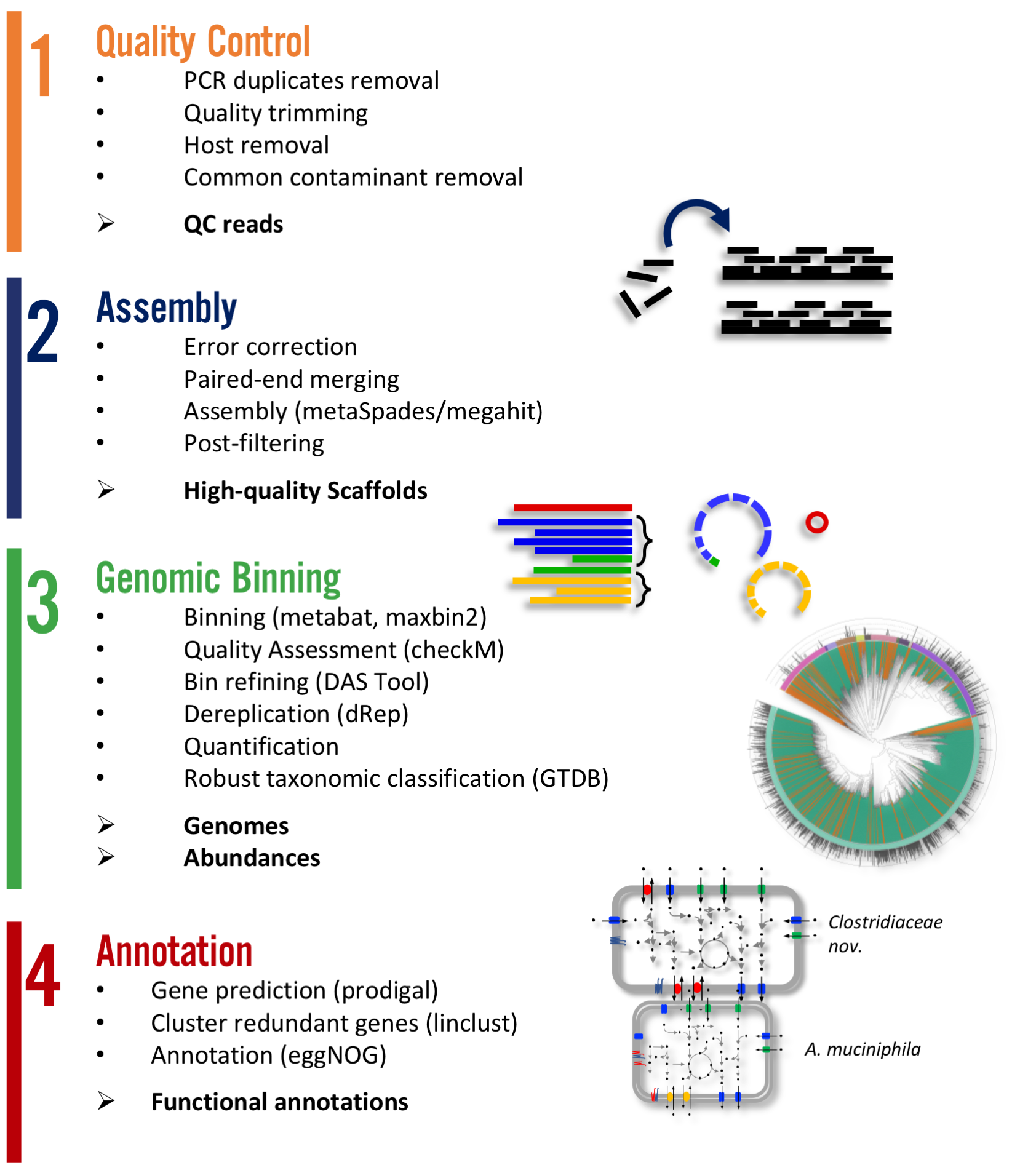
Metagenome-atlas is a easy-to-use metagenomic pipeline based on snakemake. It handles all steps from QC, Assembly, Binning, to Annotation.
You can start using atlas with three commands:
mamba install -y -c bioconda -c conda-forge metagenome-atlas={latest_version}
atlas init --db-dir databases path/to/fastq/files
atlas run all
where {latest_version} should be replaced by

https://metagenome-atlas.readthedocs.io/
ATLAS: a Snakemake workflow for assembly, annotation, and genomic binning of metagenome sequence data.
Kieser, S., Brown, J., Zdobnov, E. M., Trajkovski, M. & McCue, L. A.
BMC Bioinformatics 21, 257 (2020).
doi: 10.1186/s12859-020-03585-4
Here are some ideas I work or want to work on when I have time. If you want to contribute or have some ideas let me know via a feature request issue.
- Optimized MAG recovery (e.g. Spacegraphcats)
- Integration of viruses/plasmid that live for now as extensions
- Add statistics and visualisations as in atlas_analyze
- Implementation of most rules as snakemake wrapper
- Cloud execution
- Update to new Snakemake version and use cool reports.