


5/23/24 -> vAMPirus manuscript is out now! Check it out is Molecular Ecology Resources -- https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13978
10/23/2023 -> vAMPirus manuscript is currently in revision in Molecular Ecology Resources and there are plans to update the framework within the next year. Please note to run the vAMPirus.nf script you will not be able to use the most recent version of Nextflow (v23). You can download version 22 and it will run perfectly fine still. As always, please let us know if you run into any issues.
2/9/23 -> vAMPirus preprint has been released, check it out here: https://www.authorea.com/users/584435/articles/623635-vampirus-a-versatile-amplicon-processing-and-analysis-program-for-studying-viruses
1/20/23 -> We have created the vAMPirus Analysis Repository community on Zenodo for everyone to upload their non-read files used for their vAMPirus analyses. This is an initiative to increase shareability and reproducibility of virus amplicon sequencing analyses.
1/10/23 -> Updated code to produce the NMDS plot in the report, still not fully tested but changes should prevent errors from using deprecated code.
12/16/22 -> We noticed there were some changes in the new version of vegan which we updated to in v2.1.0. We are working to fix the issue but the reports should be generated without error, if you experience an error, you can still find the files used to generate results within the results directories. You can also use --skipReport to skip the report generation all together.
11/27/22 -> we have updated vAMPirus to v2.1.0
- New in vAMPirus version 2.1.0
- Quick intro
- Getting started
- Installing vAMPirus
- Testing vAMPirus install
- Running vAMPirus
- Who to cite
Viruses are the most abundant biological entities on the planet and with advances in next-generation sequencing technologies, there has been significant effort in deciphering the global virome and its impact in nature (Suttle 2007; Breitbart 2019). A common method for studying viruses in the lab or environment is amplicon sequencing, an economic and effective approach for investigating virus diversity and community dynamics. The highly targeted nature of amplicon sequencing allows in-depth characterization of genetic variants within a specific taxonomic grouping facilitating both virus discovery and screening within samples. Although, the high volume of amplicon data produced combined with the highly variable nature of virus evolution across different genes and virus-types can make it difficult to scale and standardize analytical approaches. Here we present vAMPirus (https://github.com/Aveglia/vAMPirus.git), an automated and easy-to-use virus amplicon sequencing analysis program that is integrated with the Nextflow workflow manager facilitating easy scalability and standardization of analyses.
"Reproducibility and research integrity are essential tenets of every scientific study and discovery. They serve as proof that an established and documented work can be verified, repeated, and reproduced.." - Diaba-Nuhoho and Amponsah-Offeh 2021 (https://tinyurl.com/279recr3)
To promote and simplify the sharing and reproduction of vAMPirus analyses we have created a Zenodo Community 'vAMPirus Analysis Repository' (zenodo.org/communities/vampirusrepo) that is meant to be a central location for all vAMPirus analyses described in a published report/preprint/manuscript. Here investigators will archive and share the non-read files in a compressed folder required to reproduce their virus amplicon analyses.
For the benefit of the field and science as a whole, we recommend uploading all non-read files needed to reproduce your analysis. For more information see the manual here: https://github.com/Aveglia/vAMPirus/blob/master/docs/HelpDocumentation.md#vampirus-analysis-repository----zenodo-community
-
Supports single-end read libraries as input.
-
Changed to have process-specific Conda evironments and Singularity/Docker containers (should help with stability).
-
Added output of R-based analyses performed during Report generation.
-
Use of Alignment Ensemble approach from musclev5 for high confidence sequence alignments
-
Added the use of Transfer Bootstrap Exptecation (TBE) in IQTREE analyses.
-
Reduced redundancy of processes and the volume of generated result files per full run (Example - read processing only done once if running DataCheck then Analyze).
-
Added further taxonomic classification of sequences using the RVDB annotation database and/or NCBI taxonomy files (see manual for more info).
-
Replaced the used of MAFFT with muscle v5 (Edgar 2021) for higher confidence virus gene alignments (see https://www.biorxiv.org/content/10.1101/2021.06.20.449169v1.full).
-
Added multiple primer pair removal to deal with multiplexed amplicon libraries.
-
ASV filtering - you can now provide a "filter" and "keep" database to remove certain sequences from the analysis
-
(EXPERIMENTAL) Added Minimum Entropy Decomposition analysis using the oligotyping program produced by the Meren Lab. This allows for sequence clustering based on sequence positions of interest (biologically meaningful) or top positions with the highest Shannon's Entropy (read more here: https://merenlab.org/software/oligotyping/ ; and below).
-
Phylogeny-based clustering ASV or AminoType sequences with TreeCluster (https://github.com/niemasd/TreeCluster; https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0221068)
-
Color nodes on phylogenetic trees based on Taxonomy or Minimum Entropy Decomposition results or TreeClustering results.
-
PCoA plots added to Analyze report if NMDS does not converge.
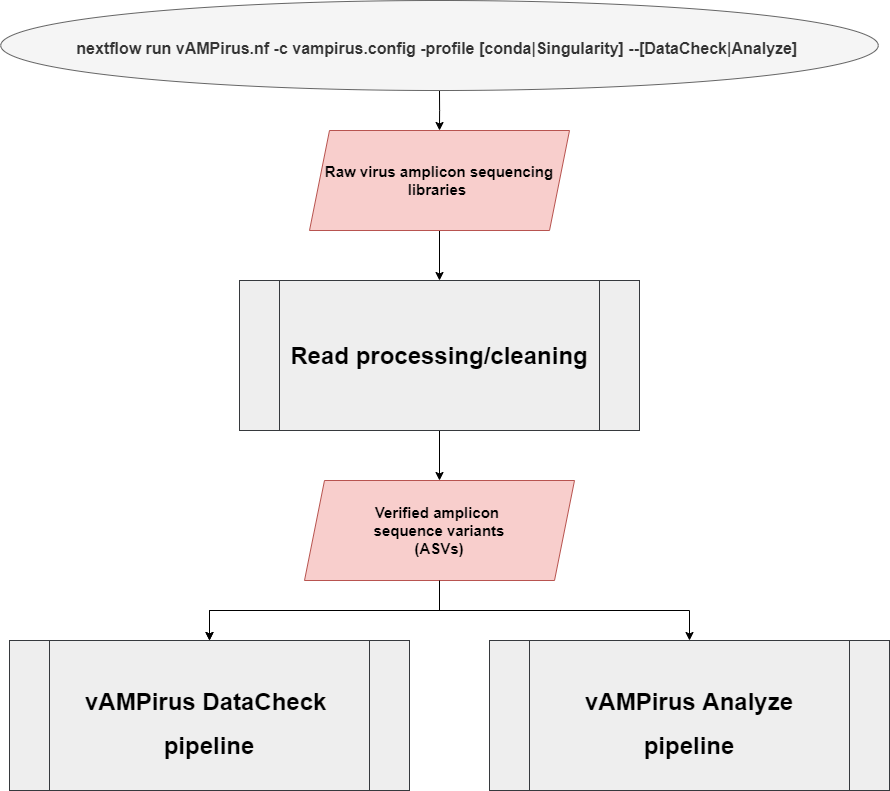
The vAMPirus program contains two different pipelines:
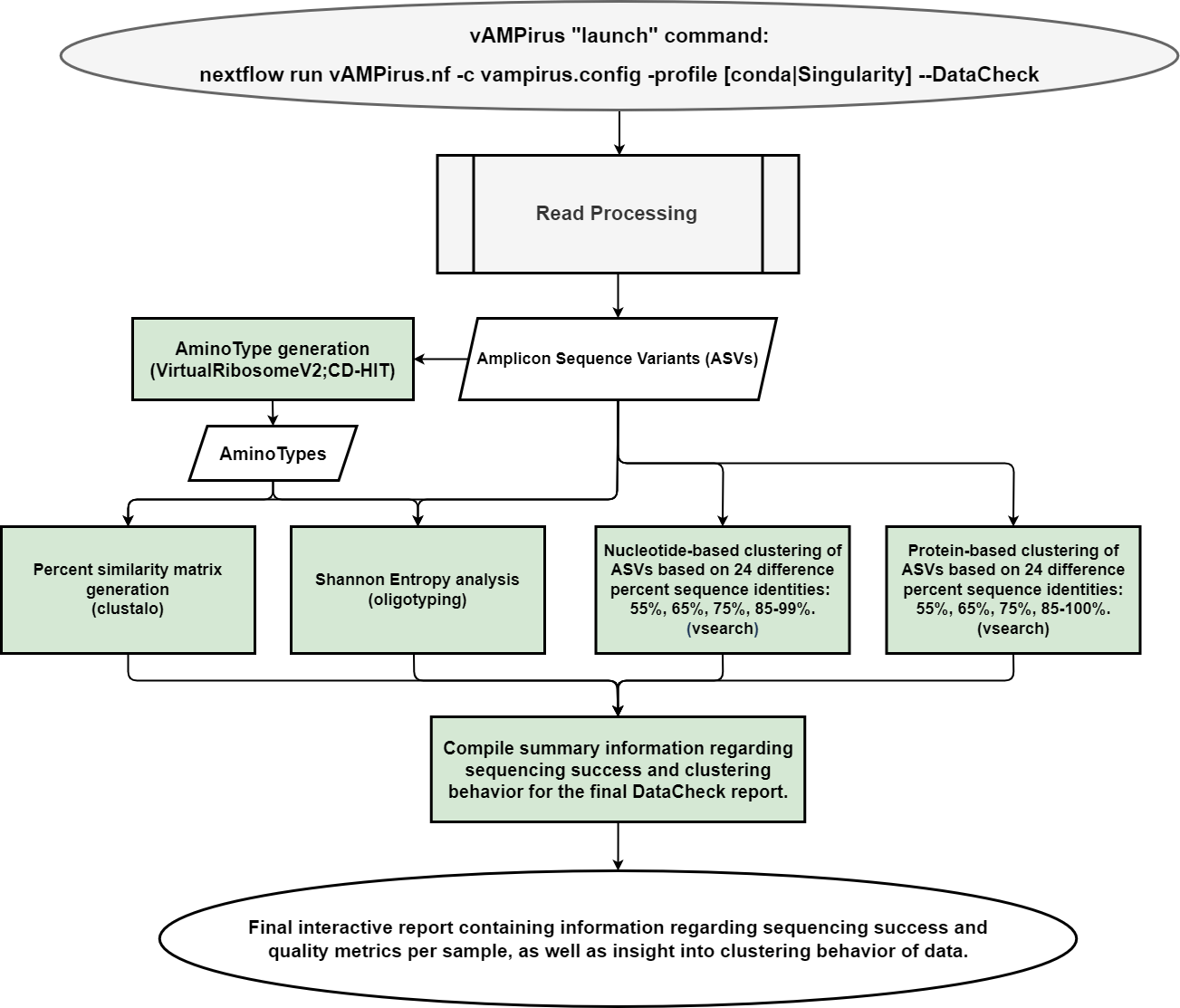
- DataCheck pipeline: provides the user an interactive html report file containing information regarding sequencing success per sample as well as a preliminary look into the clustering behavior of the data which can be leveraged by the user to inform future analyses
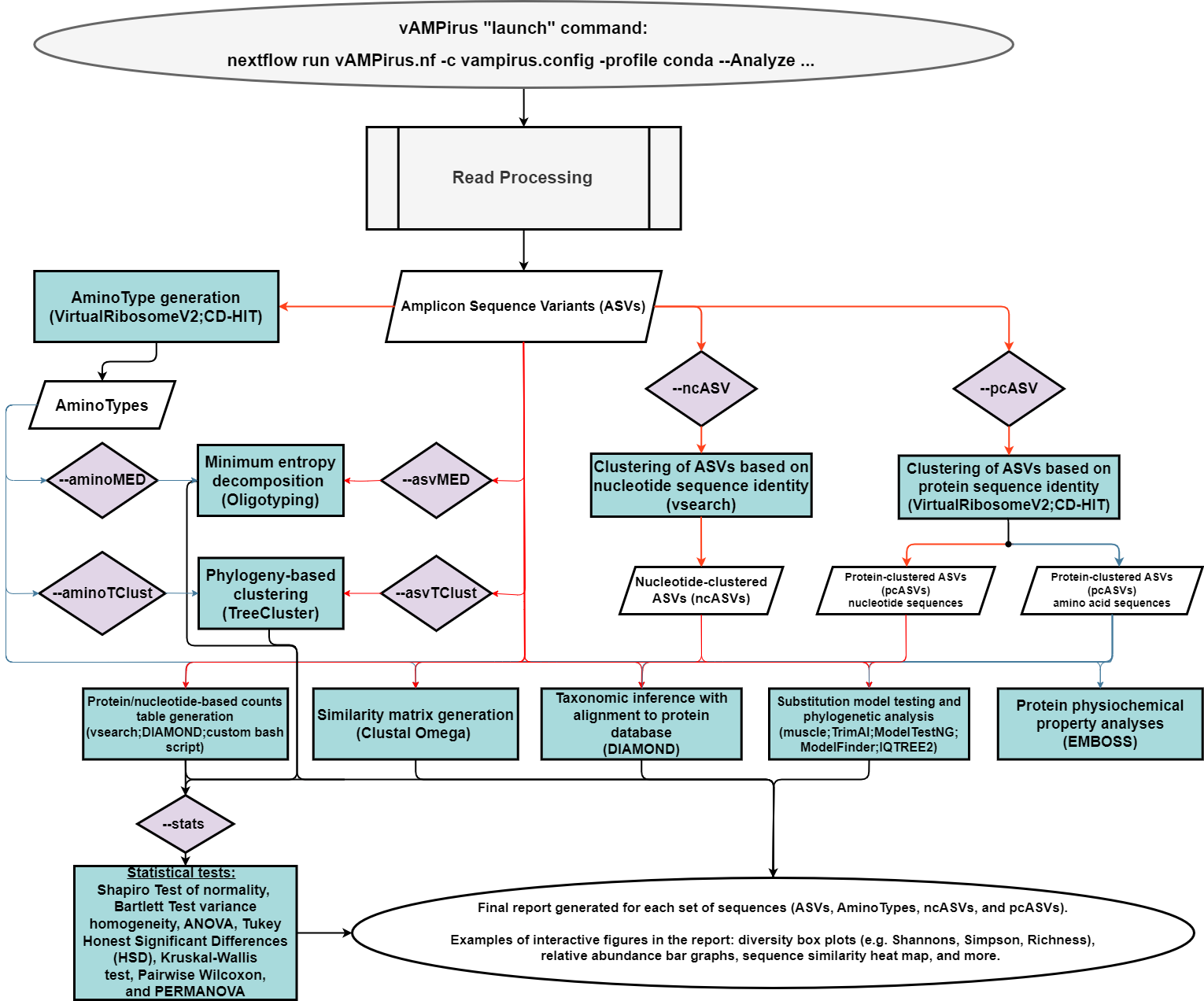
- Analyze pipeline: a comprehensive analysis of the provided data producing a wide range of results and outputs which includes an interactive report with figures and statistics. Red line represents path for nucleotide sequences, blue represents path for protein sequences.
NOTE => This is a more brief overview of how to install and set up vAMPirus, for more detail see the manual.
If you have a feature request or any feedback/questions, feel free to email vAMPirusHelp@gmail.com or you can open an Issue on GitHub.
-
Clone vAMPirus from github
-
Before launching the vAMPirus.nf, be sure to run the vampirus_startup.sh script to install dependencies and/or databases (NOTE: You will need to have the xz program installed before running startup script when downloading the RVDB database)
-
Test the vAMPirus installation with the provided test dataset (if you have ran the start up script, you can see EXAMPLE_COMMANDS.txt in the vAMPirus directory for test commands and other examples)
-
Edit parameters in vampirus.config file
-
Launch the DataCheck pipeline to get summary information about your dataset (e.g. sequencing success, read quality information, clustering behavior of ASV or AminoTypes)
-
Change any parameters in vampirus.config file that might aid your analysis (e.g. clustering ID, maximum merged read length, Shannon entropy analysis results)
-
Launch the Analyze pipeline to perform a comprehensive analysis with your dataset
-
Explore results directories and produced final reports
New in v3.0.0 the dependencies of vAMPirus will be set up by Nextflow upon the initial launch of a vAMPirus pipeline regardless if you plan to use Conda or Singularity/Docker.
Run the start up script to set paths automatically in the configuration file, install Conda, and install any databases.
vAMPirus has been set up and tested on Windows 10 using Ubuntu Sandbox (https://wiki.ubuntu.com/WSL) which is a feature of Windows 10 - See Windows Subsystem for Linux -> https://docs.microsoft.com/en-us/windows/wsl/about
All you will need to do is set up the subsystem with whatever flavor of Linux you favor and then you can follow the directions for installation and running as normal.
Search for Linux in the Microsoft store -> https://www.microsoft.com/en-us/search?q=linux
For more detail see the manual.
If you plan to run vAMPirus on a Mac computer, it is recommended that you set up a virtual environment and use Singularity with the vAMPirus Docker image to run your analyses.
You can try to run directly on your system, but there may be errors caused by differences between Apply and GNU versions of tools like "sort".
For more detail see the manual.
Clone the most recent version of vAMPirus from GitHub using:
git clone https://github.com/Aveglia/vAMPirus.git
OR you can download the most recent stable release vAMPirus v2.0.0 (Polymerase I) by using:
wget https://github.com/Aveglia/vAMPirus/archive/refs/tags/v2.0.0.tar.gz
vAMPirus is integrated with Nextflow (https://www.Nextflow.io) which relies on Java being installed on your system.
If you know you do not have it installed, see here for instructions on installing Java for different operating software's -> https://opensource.com/article/19/11/install-java-linux ; for Debian https://phoenixnap.com/kb/how-to-install-java-ubuntu
If you are unsure, you can check using:
which java
or
java -version
The output from either of those commands should let you know if you have Java on your system.
You will also need to decide if you plan to use a container engine like Docker (https://www.docker.com/) or Singularity (https://singularity.lbl.gov/) or the package manager Conda (https://docs.conda.io/en/latest/).
The startup script provided in the vAMPirus program directory will install Conda for you if you tell it to (see below), however, unless you plan to run in the Vagrant VM as described above, you will need to install Docker or Singularity before running vAMPirus.
To set up and install vAMPirus dependencies (e.g., Conda, Nextflow), simply move to the vAMPirus directory and run the vampirus_startup.sh script.
cd ./vAMPirus; bash vampirus_startup.sh -h
You can make the vampirus_startup.sh scrip an exectuable with -> chmod +x vampirus_startup.sh ; ./vampirus_startup.sh
The start up script will check your system for Nextflow and Anaconda/Miniconda (can be skipped) and if they are not present, the script will ask if you would like to install these programs. If you answer with 'y', the script will install the missing programs and the installation is complete.
You can also use the startup script to install different databases to use for vAMPirus analyses, these include:
- NCBIs Viral protein RefSeq database
- The proteic version of the Reference Viral DataBase (RVDB) (See https://f1000research.com/articles/8-530)
- The complete NCBI NR protein database
To use the vampirus_startup.sh script to download any or all of these databases listed above you just need to use the "-d" option and you can download the NCBI taxonomy files with the option "-t" (See below).
If we take a look at the vampirus_startup.sh script usage:
General execution:
vampirus_startup.sh -h [-d 1|2|3|4] [-s] [-t]
Command line options:
[ -h ] Print help information
[ -d 1|2|3|4 ] Set this option to create a database directiory within the current working directory and download the following databases for taxonomy assignment:
1 - Download only the proteic version of the Reference Viral DataBase (See the paper for more information on this database: https://f1000research.com/articles/8-530)
2 - Download only NCBIs Viral protein RefSeq database
3 - Download only the complete NCBI NR protein database
4 - Download all three databases
[ -s ] Set this option to skip conda installation and environment set up (you can use if you plan to run with Singularity and the vAMPirus Docker container)
[ -t ] Set this option to download NCBI taxonomy files needed for DIAMOND to assign taxonomic classification to sequences (works with NCBI type databases only, see manual for more information)
For example, if you would like to install Nextflow, download NCBIs Viral Protein RefSeq database, the NCBI taxonomy files to use DIAMOND taxonomy assignment feature, and check/install conda, run:
bash vampirus_startup.sh -d 2 -t
and if we wanted to do the same thing as above but skip the Conda check/installation, run:
bash vampirus_startup.sh -d 2 -s
NOTE -> if you end up installing Miniconda3 using the script you might have to close and re-open the terminal window after everything is completed.
**NEW in version 2.0.0 -> the startup script will automatically download annotation information from RVDB to infer Lowest Common Ancestor (LCA) information for hits during taxonomy assignment. You can also use "-t" to download NCBI taxonomy files to infer taxonomy using the DIAMOND taxonomy classification feature.
The startup script will generate a text file (STARTUP_HELP.txt) that has instructions and example commands to test the installation.
NOTE => If using Singularity, when you run the test command calling for singularity (-profile singularity) Nextflow will set up the dependencies.
Launch commands for testing (you do not need to edit anything in the config files for test commands):
`/path/to/nextflow run /path/to/vAMPirus.nf -c /path/to/vampirus.config -profile conda,test --DataCheck -resume`
OR
`nextflow run vAMPirus.nf -c vampirus.config -profile singularity,test --DataCheck -resume`
`/path/to/nextflow run /path/to/vAMPirus.nf -c /path/to/vampirus.config -profile conda,test --Analyze -resume`
OR
`nextflow run vAMPirus.nf -c vampirus.config -profile singularity,test --Analyze -resume`
If you done the setup and confirmed installation success with the test commands, you are good to get going with your own data. Before getting started edit the configuration file with the parameters and other options you plan to use.
Here are some example vAMPirus launch commands:
Example 1. Launching the vAMPirus DataCheck pipeline using conda and Shannon Entropy Analysis on ASVs and AminoTypes
`nextflow run vAMPirus.nf -c vampirus.config -profile conda --DataCheck --asvMED --aminoMED`
Example 2. Launching the vAMPirus DataCheck pipeline using Singularity and multiple primer removal with the path to the fasta file with the primer sequences set in the launch command
nextflow run vAMPirus.nf -c vampirus.config -profile singularity --DataCheck --multi --primers /PATH/TO/PRIMERs.fa
Example 3. Launching the vAMPirus DataCheck pipeline with primer removal by global trimming of 20 bp from forward reads and 26 bp from reverse reads
nextflow run vAMPirus.nf -c vampirus.config -profile conda --DataCheck --GlobTrim 20,26
Example 4. Launching the vAMPirus Analyze pipeline with singularity with ASV and AminoType generation with all accesory analyses (taxonomy assignment, EMBOSS, IQTREE, statistics)
`nextflow run vAMPirus.nf -c vampirus.config -profile singularity --Analyze --stats`
Example 5. Launching the vAMPirus Analyze pipeline with conda to perform multiple primer removal and protein-based clustering of ASVs, but skip most of the extra analyses
nextflow run vAMPirus.nf -c vampirus.config -profile conda --Analyze --pcASV --skipPhylogeny --skipEMBOSS --skipTaxonomy --skipReport
Example 6. Launching vAMPirus Analyze pipeline with conda to produce only ASV and AminoType-based results with Shannon Entropy Analyses with the nodes on produced phylogenies colored based on taxnomy hit
`nextflow run vAMPirus.nf -c vampirus.config -profile conda --Analyze --asvMED --aminoMED --nodeCol TAX --stats`
If an analysis is interrupted, you can use Nextflows "-resume" option that will start from the last cached "check point".
For example if the analysis launched with the command from Example 6 above was interrupted, all you would need to do is add the "-resume" to the end of the command like so:
`nextflow run vAMPirus.nf -c vampirus.config -profile conda --Analyze --asvMED --aminoMED --nodeCol TAX --stats -resume`
If you do use vAMPirus for your analyses, please cite the following ->
-
vAMPirus - Veglia, A. J., Rivera-Vicéns, R. E., Grupstra, C. G. B., Howe-Kerr, L. I., & Correa, A. M. S. (2024). vAMPirus: A versatile amplicon processing and analysis program for studying viruses. Molecular Ecology Resources, 00, e13978. https://doi.org/10.1111/1755-0998.13978
-
DIAMOND - Buchfink B, Xie C, Huson DH. (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12(1):59-60. doi:10.1038/nmeth.3176
-
FastQC - Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
fastp - Chen, S., Zhou, Y., Chen, Y., & Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 34(17), i884-i890.
-
Clustal Omega - Sievers, F., Wilm, A., Dineen, D., Gibson, T.J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Söding, J. and Thompson, J.D., 2011. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular systems biology, 7(1), p.539.
-
IQ-TREE - Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., & Lanfear, R. (2020). IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37(5), 1530-1534.
-
ModelTest-NG - Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., & Flouri, T. (2020). ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Molecular biology and evolution, 37(1), 291-294.
-
muscle v5 - R.C. Edgar (2021) "MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping" https://www.biorxiv.org/content/10.1101/2021.06.20.449169v1.full.pdf
-
vsearch - Rognes, T., Flouri, T., Nichols, B., Quince, C., & Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ, 4, e2584.
-
BBMap - Bushnell, B. (2014). BBTools software package. URL http://sourceforge. net/projects/bbmap.
-
trimAl - Capella-Gutiérrez, S., Silla-Martínez, J. M., & Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics, 25(15), 1972-1973.
-
CD-HIT - Fu, L., Niu, B., Zhu, Z., Wu, S., & Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28(23), 3150-3152.
-
EMBOSS - Rice, P., Longden, I., & Bleasby, A. (2000). EMBOSS: the European molecular biology open software suite.
-
seqtk - Li, H. (2012). seqtk Toolkit for processing sequences in FASTA/Q formats. GitHub, 767, 69.
-
UNOISE algorithm - R.C. Edgar (2016). UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing, https://doi.org/10.1101/081257
-
Oligotyping - A. Murat Eren, Gary G. Borisy, Susan M. Huse, Jessica L. Mark Welch (2014). Oligotyping analysis of the human oral microbiome. Proceedings of the National Academy of Sciences Jul 2014, 111 (28) E2875-E2884; DOI: 10.1073/pnas.1409644111
-
Balaban M, Moshiri N, Mai U, Jia X, Mirarab S (2019). "TreeCluster: Clustering biological sequences using phylogenetic trees." PLoS ONE. 14(8):e0221068. doi:10.1371/journal.pone.0221068
-
Wernersson R. Virtual Ribosome--a comprehensive DNA translation tool with support for integration of sequence feature annotation. Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W385-8. doi: 10.1093/nar/gkl252. PMID: 16845033; PMCID: PMC1538826.